Apa yang pertama kali terbayangkan ketika kalian mendengar kata “kimia”? Pasti laboratorium, reaksi, dan simbol-simbol atom, bukan? Kali ini kita akan memandang kimia dari sudut yang berbeda. Pernahkah kalian membayangkan apa sih yang sebenarnya terjadi ketika kita melakukan reaksi? Bagaimana ikatan antaratom itu dapat putus kemudian terbentuk?
Secara eksperimen, pengamatan apa yang sedang terjadi ketika reaksi kimia berlangsung sangat sulit dilakukan. Mengapa? Karena sebagian besar reaksi kimia terjadi dalam skala waktu yang sangat cepat. Hal ini yang lumayan sulit diikuti oleh alat instrumen kimia. Padahal, apabila kita dapat mengetahui apa yang terjadi selama reaksi kimia berlangsung, kita dapat menyusun mekanisme reaksi yang terjadi, memahami bagaimana sebuah reaksi kimia terjadi, dan pada akhirnya dapat mendesain material baru yang lebih baik. Untuk itulah para ilmuwan berpikir bagaimana membawa “eksperimen laboratorium” ke “eksperimen komputer”.
Apa sih “eksperimen komputer” itu? Eksperimen komputer merupakan istilah lain untuk simulasi dinamika molekular. Simulasi dinamika molekular artinya kita meniru sistem kimia nyata, membawanya ke dalam komputer yang tujuannya adalah mengamati bagaimana pergerakan molekul, bagaimana interaksi antarmolekul, bagaimana sebuah ikatan dapat putus kemudian terjadi lagi selama waktu tertentu dalam rentang ps (piko detik, 1 ps = 10-12 s).
Mengapa hal ini bisa dilakukan? Ini ada hubungannya dengan hukum kedua Newton,
Gaya (F) = massa (m) x percepatan (a)
Berdasarkan persamaan ini, kita bisa mengetahui posisi molekul pada “masa lalu”, “masa kini”, dan “masa depan” dengan cara melakukan integrasi persamaan Newton menggunakan algoritma integrasi tertentu. Prinsip yang kedua adalah hipotesis ergodik yang artinya “rata-rata hasil pengukuran untuk banyak molekul dalam waktu yang singkat akan sama dengan hasil pengukuran untuk molekul yang lebih sedikit tetapi dalam waktu yang lebih lama”. Dengan prinsip ini, kita tidak perlu melakukan simulasi untuk miliaran partikel, tetapi cukup ratusan hingga ribuan partikel saja. Lagi pula, perangkat komputasi yang tersedia tidak memungkinkan untuk melakukan integrasi persamaan Newton hingga milyaran partikel.
Lalu, bagaimana caranya agar simulasi dinamika molekular dapat meniru apa yang terjadi dalam eksperimen? Nah, ada beberapa hal yang perlu kita pertimbangkan. Kita mulai dari yang sederhana yaitu periodisitas. Periodisitas berarti kotak simulasi kita harus berulang ke segala arah hingga jarak yang tidak terhingga. Dengan demikian, kita akan mendapatkan sifat bulky atau meruah dan efek permukaan akan hilang. Konsep periodisitas ini diikuti dengan penetapan batas kapan interaksi antarpartikel akan dihitung atau diabaikan. Hal ini bertujuan agar kita tidak perlu menghitung interaksi antara semua partikel, cukup interaksi antarpartikel dalam radius jarak tertentu yang kita hitung, selebihnya kita anggap mereka tidak berinteraksi atau energi interaksi mereka sama dengan 0.
Konsep penting lain dalam simulasi dinamika molekular adalah ensemble. Jika teman-teman melakukan reaksi di laboratorium, akan ada variabel yang dijaga konstan, baik itu tekanan, volume, dan temperatur. Nah, dalam simulasi dinamika molekular kita bisa menjaga variabel-variabel tersebut konstan selama simulasi. Artinya, hasil perhitungan kita dilakukan dalam kondisi ada variabel yang dijaga tetap. Jika kita menjaga agar jumlah partikel (N), tekanan (P), dan suhu (T) tetap, hal tersebut dinamakan ensemble NPT. Apabila yang dijaga tetap adalah jumlah partikel (N), volume (V), dan suhu (T), hal tersebut dinamakan ensemble NVT.
Untuk mendeskripsikan interaksi antarmolekul, kita memerlukan sesuatu yang disebut potensial pasangan. Potensial pasangan ini nanti yang mengatur bagaimana interaksi antarmolekul sehingga mirip dengan interaksi mereka di dalam sistem kimia yang nyata. Bisa dibilang potensial pasangan inilah yang menjadi faktor penting dari simulasi dinamika molekular karena tanpa itu kita tidak akan bisa menggambarkan interaksi antarmolekul dengan benar.
Dalam mendeskripsikan interaksi antaratom/partikel untuk sistem yang sangat kompleks seperti protein, kita memerlukan medan gaya. Medan gaya merupakan kumpulan data berisi panjang ikatan, sudut, momen gaya, bahkan interaksi elektrostatis antarpartikel. Setiap medan gaya dibuat spesifik tergantung dari mana data tersebut dihasilkan. Medan gaya untuk protein akan berbeda dengan medan gaya untuk rantai hidrokarbon. Medan gaya yang sering digunakan untuk memodelkan protein antara lain AMBER dan CHARMM.
Apakah potensial pasangan ini sebuah keharusan? Tidak, potensial ini digunakan ketika kita melakukan simulasi dinamika molekular klasik yang menggunakan hukum kedua Newton. Apabila basis simulasi kita tidak berdasarkan pada mekanika klasik melainkan mekanika kuantum, kita tidak lagi memerlukan potensial pasangan dan medan gaya.

Kelemahan penggunaan medan gaya adalah kita tidak dapat menjelaskan terjadinya pembentukan atau pemutusan ikatan antaratom karena tidak ada deskripsi tersebut di dalam medan gaya. Untuk mengatasi hal itu, sistem simulasi kita bagi menjadi dua, yakni bagian mekanika kuantum (yang tidak memerlukan medan gaya) dan bagian mekanika molekular/klasik (yang memerlukan medan gaya). Ide ini pertama kali dicetuskan oleh Martin Karplus, Michael Levitt dan Arieh Warshel, ketiganya merupakan penerima hadiah Nobel bidang kimia tahun 2013. Simulasi dinamika molekular dengan metode ini dikenal dengan nama simulasi dinamika molekular mekanika kuantum/mekanika molekul (MK/MM).
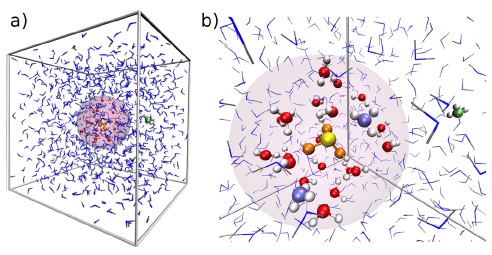
Gambar (b): gambaran lebih detail daerah mekanika kuantum (Canaval dkk., 2014).
Konsep MK/MM sangat bermanfaat karena bagian yang menjadi pusat perhatian dihitung menggunakan metode mekanika kuantum (yang lebih akurat daripada mekanika molekular) dan bagian yang lebih besar dihitung menggunakan mekanika molekular (yang lebih cepat daripada mekanika kuantum) sehingga dapat menghemat sumber daya dan waktu komputasi.
Karena simulasi dinamika molekular merupakan eksperimen komputer, maka kita perlu perangkat komputasi yang mumpuni, tidak bisa hanya menggunakan notebook atau netbook. Perangkat seperti apa? Yang jelas kita butuh prosesor dengan core yang banyak dan cepat. Semakin banyak core artinya semakin banyak job yang bisa didistribusikan ke dalam core sehingga pekerjaan kita akan lebih cepat selesai. Namun, yang perlu diingat adalah antara waktu yang diperlukan agar simulasi selesai tidak dapat berbanding lurus dengan jumlah prosesor.
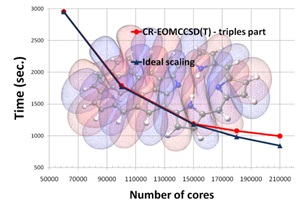
Jika teman-teman ingin mencoba melakukan simulasi, teman-teman dapat menggunakan spesifikasi komputer gaming yang sering disarankan di majalah komputer. Daripada beli komputer mahal hanya untuk main game, lebih baik digunakan untuk hal yang bermanfaat. Dalam skala riset, simulasi sering dilakukan menggunakan kluster komputer, yaitu sebuah perangkat keras yang terdiri dari ratusan-ribuan prosesor yang saling terhubung melalui interkoneksi yang sangat cepat. Misalnya di LIPI, terdapat kluster komputer yang dapat diakses untuk melakukan riset tentang simulasi dinamika molekular (http://grid.lipi.go.id/).
Selain prosesor, kita juga perlu memilih RAM dan hard disk dengan kapasitas yang besar. Penggunaan hard disk dengan kapasitas yang besar sangat disarankan karena file-file yang dihasilkan oleh simulasi bisa menjadi sangat besar tergantung dari sistem simulasi dan waktu simulasi yang akan kita jalankan. Dalam era sekarang, penggunakan GPU (Graphical Processing Unit) juga mempercepat waktu komputasi secara signifikan. Hal ini dikarenakan GPU memiliki core yang puluhan kali lebih banyak daripada prosesor sehingga dapat melakukan simulasi dengan lebih cepat.
Sayangnya, tidak semua GPU dapat digunakan untuk simulasi, hanya beberapa versi GPU tertentu, terutama GPU model baru yang dapat digunakan. Kelemahan lain dari GPU adalah mereka lebih gampang rusak daripada CPU apabila digunakan secara terus menerus tanpa henti. Meski demikian, simulasi dinamika molekular untuk sistem biomolekul sangat terbantu dengan penggunaan GPU karena waktu komputasi bisa dipersingkat.
Bahan bacaan:
- Canaval, L., Lutz, O., Weiss, A., 2014. A Dissociative Quantum Mechanical/Molecular Mechanical Molecular Dynamics Simulation and Infrared Experiments Reveal Characteristics of the Strongly Hydrolytic. Chem.
- Jitonnom, J., Lee, V.S., Nimmanpipug, P., Rowlands, H.A., Mulholland, A.J., 2011. Quantum Mechanics/Molecular Mechanics Modeling of Substrate-Assisted Catalysis in Family 18 Chitinases: Conformational Changes and the Role of Asp142 in Catalysis in ChiB. Biochemistry, 50, 4697–4711.
Penulis:
Niko Prasetyo, alumnus S2 Kimia Universitas Gadjah Mada.
Kontak: neax.debian(at)gmail(dot)com.


